Introduction
If you have found your way here, you are probably already aware of the research into so-called “glass batteries” led by Maria Helena Braga and recent Nobel laureate John Goodenough.
What this group have presented over the last few years has been widely touted in technology magazines and by the team themselves as the long awaited game-changer in battery technology: safe, high energy density, high power, wide operating temperature window, long cycle life, and constructed using only cheap, environmentally friendly materials. However, this work has been met with deep criticism in the battery research community, for wildly exaggerated claims, weak analysis, and highly questionable research practices.
When this work first reached public attention, I laid out some of my technical criticisms of two of their research papers in two previous posts, here and here. It has now been close to two years since my last post, but Braga, Goodenough and other co-authors have published a number of new papers since then — many of them now open access, where previously they were largely paywalled — and now and again this work finds fresh attention in the press.
In this post, I am most interested in their most recent paper, “Performance of a ferroelectric glass electrolyte in a self-charging electrochemical cell with negative capacitance and resistance”. There’s much to say about this paper, as we will see, but I will also try and connect the dots from my earlier posts. As with my previous two, I hope to dig into some of the technical details to try and make sense of what is being presented, and where some of the deeper problems lie.
I know that many readers of my earlier posts are not trained in the dark art of batteries and that this can be difficult to follow. For that, I am sorry - this work is difficult to unpack even for those familiar with the field. My hope is to cut down on the field-specific jargon and write as accessibly as possible (and for where I don’t, you are welcome to ask questions in the comments). But that said, I consider it important to try and engage with this work on a deeper level, and try and explain both what it is trying to argue and where the thinking goes astray. For those with the patience to make it to the end, thank you in advance for reading.
An evolving body of work
Let’s recap. This thread of research on “glass batteries”, headed by Braga, Goodenough and other researchers (hereafter referred to as Braga et al. or “the authors”), now spans no less than nine research papers (plus several related patents). For the reader’s interest, here those papers:
- Novel Li3ClO based glasses with superionic properties for lithium batteries, J. Mater. Chem. A (2014)
- Glass-amorphous alkali-ion solid electrolytes and their performance in symmetrical cells, Energy Environ. Sci. (2016)
- Electric Dipoles and Ionic Conductivity in a Na+ Glass Electrolyte, J. Electrochem. Soc. (2016)
- Alternative strategy for a safe rechargeable battery, Energy Environ. Sci. (2017)
- Nontraditional, Safe, High Voltage Rechargeable Cells of Long Cycle Life, J. Am. Chem. Soc. (2018)
- Extraordinary Dielectric Properties at Heterojunctions of Amorphous Ferroelectrics, J. Am. Chem. Soc. (2018)
- Low-Temperature Performance of a Ferroelectric Glass Electrolyte, ACS Appl. Energy Mater. (2019)
- Thermodynamic considerations of same-metal electrodes in an asymmetric cell, Materials Theory (2019)
- Performance of a ferroelectric glass electrolyte in a self-charging electrochemical cell with negative capacitance and resistance, Appl. Phys. Reviews (2020)
Since my last post on this almost two years ago, the authors have published a further four (6 — 9), including the one we will discuss here.
By now, therefore, this is a rather developed body of work, with some common themes running through the work in general. But more than that — we have had a little bit of related work done by other scientists which shed some light on the situation.
What is this latest paper about?
This is, as best I can work out, how it goes:
Braga et al. state that they have developed a new battery which charges itself — something that would be revolutionary in energy storage. They have done this before, of course, but this time the electrodes are not made with typical battery materials — only base metals (copper, aluminium, zinc, and so on). Not only do they “self-charge”, but they do so in semi-regular bursts (they oscillate). The authors explain that this behaviour is due to effects of “negative resistance and capacitance”, not previously observed simultaneously, supposedly arising from the ferroelectric (more on this word later) properties of their electrolyte — the same glass electrolyte they have been working with for the last few years. To explain this, they borrow from current theory on so-called negative capacitance in ferroelectric materials relevant to semiconductor physics (i.e., ferroelectric field effect transistors). Therefore, it would seem, bringing the understanding of this field to energy storage devices.
I am not especially familiar with field-effect transistors, but I will touch on this briefly to try and put it in to some sort of context. However, as we will see in the remainder of this post, I think the whole exercise is like trying to analyse a Jackson Pollock painting from the perspective of the realist movement (sorry for the art metaphor, I couldn’t think of a better one).
The electrolyte is almost certainly not a ferroelectric glass of extraordinary properties, it is a wet mush of different salts
One of the big problems I highlighted in my first “glass battery” post is that the composition and structure of the electrolyte, from which many of its supposedly remarkable properties are derived, is never shown in any convincing sense — and that it was very unlikely to be what the authors claimed the composition to be, namely a Li3OCl glass with a small amount of Li exchanged for Ba. I reasoned that such a material, if it could be prepared at all, should be extremely susceptible to reaction with water, and would have to be kept exceptionally dry in order to have the properties they were claiming. However, they use water as a solvent for the synthesis, and do most of the material handling in air, where traces of water are unavoidable. So the dryness and purity of the material was doubtful from the start.
Since then, researchers at the Technical University of Graz in Austria have repeated the preparation of this material and carried out impressively rigorous and careful characterisation work to identify its true composition and structure — repeating Braga et al’s exact method. As expected, they found, convincingly, that the material they synthesise contains a substantial amount of hydrogen — from the water — forming instead something closer to Li2(OH)Cl. Even then, they found this compound begins decomposing immediately in contact with air, forming compounds such as lithium carbonate (Li2CO3) and, crucially, hydrated lithium chloride (LiCl.xH2O) - a compound which can form a glass with a very high conductivity. On this basis, then, it would seem that the conductive component in the “Braga glass” is LiCl.xH2O, mixed in with other impurities, and not the Ba-doped Li3OCl they assumed it to be.
This is crucial: high conductivity aside, the presence of free chloride ions and water means a lot of unwanted side reactions of the electrolyte in any lithium battery system — the sort of side reactions I previously argued were almost certainly taking place and were responsible for much of the supposedly unprecedented device performance. Removing traces of water is crucial in today’s Li-ion batteries.
Not only this — it also makes the interpretation of results in terms of the “ferroelectric” character of the electrolyte essentially unsupportable, since none of the compounds that are actually found in it are ferroelectric. And the authors have not, as far as I know, presented any results that actually show that the material behaves as a ferroelectric. But there’s more…
Some record-breaking properties are thanks to basic errors of calculation
This paper states, as do the previous papers, that the “glass” has an enormous dielectric constant - 106 - 107, which would be at least an order of magnitude larger than any other known material. How can it be so large? Well, because they have been calculated wrongly. Thankfully, in this paper, as with some of the others, the calculation is presented clearly enough that the error is easy to spot. But to explain the error, I need to give a bit of background information.
The problem stems from the fact that the material, whatever it’s actually made of, is not a pure dielectric - it is an ionic conductor. The distinction is crucial. If you take a thin layer of a dielectric or an ionic conductor, and sandwich it between two electrodes, you have a capacitor. You can pass current through it and it will store (or release some amount of energy through the accumulation of charges on the surfaces of the electrodes. Dielectrics allow this to happen through the alignment of dipoles within the material. The more the dipoles can be polarised, the higher the dielectric constant (a measure of how far a material can be polarised), and the more charge can be stored on an electrode at a given voltage. Ionic conductors are also dielectrics (they have dipoles), but they also have mobile charges — ions like Li+ which move freely independently of the dipoles in the host material.
Now, this gives rise to a phenomenon called electric double layer capacitance (EDLC) — a result of a process where an electrode and electrolyte with different surface energy level are brought into contact, and there is an energy difference that needs to be equalised — this energy difference is stored in the EDLC.
The distinction is crucial, because this EDLC adds to the dielectric capacitance, and it is very large by comparison. Supercapacitors make use of EDLC to reach very high capacitances, but at the cost of a limited voltage range — because at too high voltages, the electrolyte breaks down.
This all means that it is very difficult to correctly measure the dielectric constant of an ionic conductor. If you don’t take it into account, and just try and calculate the dielectric constant from the total capacitance, you end up with unrealistically huge numbers. And that’s what Braga et al. have done.
Let’s take for example the size and thickness of the electrolyte Braga et al report - a 6.25 cm2 electrode and 1.5 mm thick. If we imagine that we have an EDLC with 20 µF/cm2 (a very typical number), for such a cell with two of the same electrodes has a total capacitance of 62.5 µF (it’s two EDLCs in series). Plugging these into the parallel plate equation gives us 1.7 x 107 - one order of magnitude lower than the 2 x 108 Braga et al. find. Add in any effects of surface roughness (increasing area), plus additional side reactions providing extra current (which can be seen in the data), can easily explain the difference.
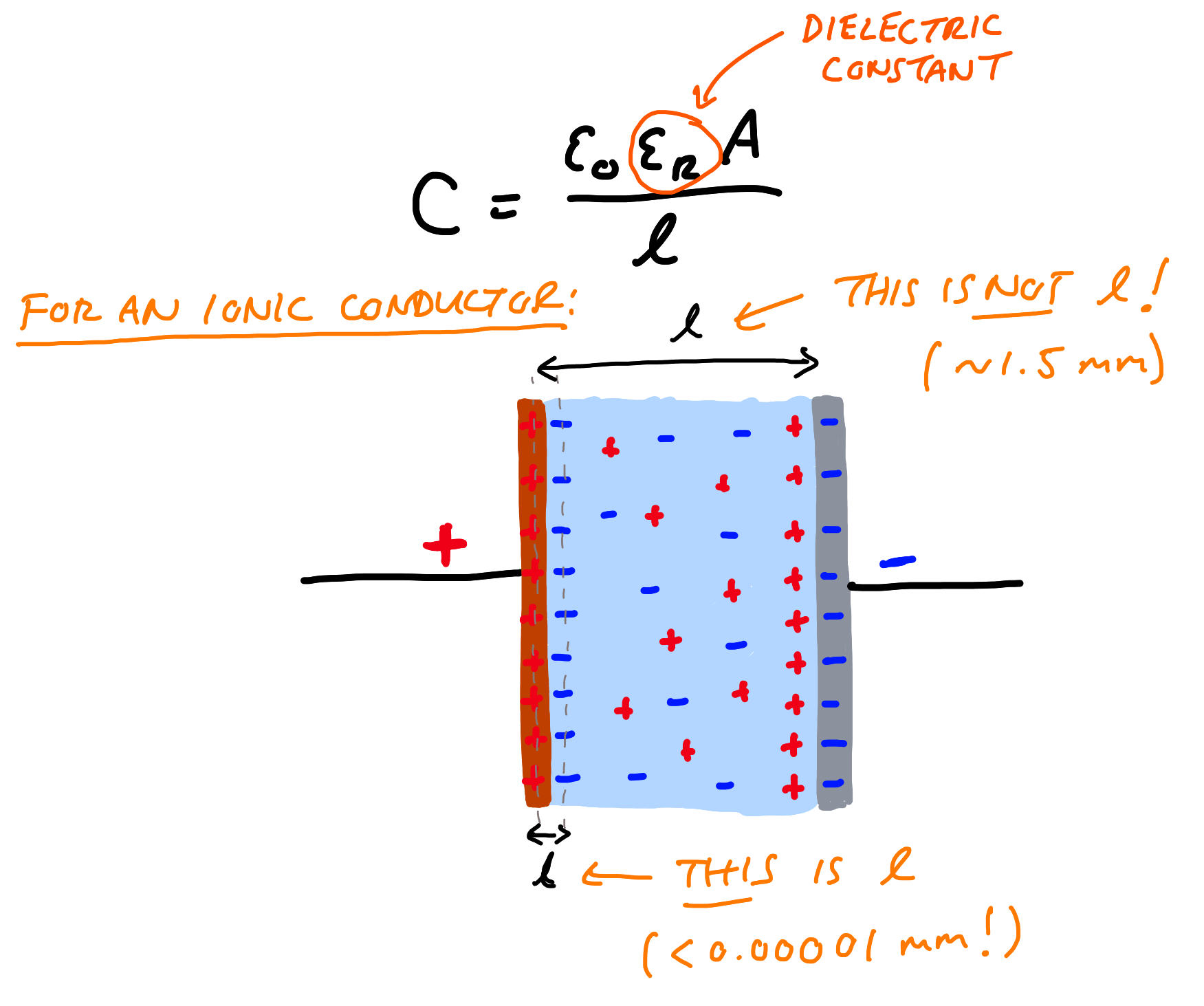
The mistake here, if it wasn’t already clear, is that to get such huge numbers requires treating something that is exclusively a surface phenomenon as a bulk property of a material - and treating a process that isn’t dielectric capacitance as if it is. The only thing more remarkable than such a basic error being made is said error slipping through peer review not just once, but several times in journals of high “prestige”. If they get this aspect of capacitance wrong, then what else?
Negative capacitance?
Ferroelectric materials are a little out of my expertise, but the word has come up so often already I can’t really avoid it — so here goes. A ferroelectric is a material which exhibits a spontaneous polarisation (alignment of dipoles), which can be switched by application of an appropriate electric field (i.e., a voltage). After the applied voltage/electric field is removed, the material stays in the state it’s in. For this reason, they’re useful for devices such as transistors, because they can keep one or the other state (or — on, or off) without needing to consume any energy to stay in a particular state.
Now, as far as I understand it — bear with me — because of the spontaneous nature of the polarisation, as they are switched from one polarisation direction to the other, they are in an unstable state during the transition. This gives rise to a phenomenon where the local electric field in the material is negative with respect to the external electric field, and a plot of polarisation vs voltage for the system becomes S-shaped (i.e., a region appears where the slope becomes negative, when ordinarily this is positive), indicating the so-called “negative capacitance” (NC) phenomenon. However, the overall capacitance in the system is still positive, otherwise it would violate the laws of thermodynamics. The material is unstable while it is in this negative capacitance region and will prefer to settle in one configuration or the other after a very short time. Nonetheless, this characteristic is of current interest in physics and ultimately electrical engineering since it gives rise to some useful electrical characteristics (not self-charge or self-cycling though…).
How this is connected to Braga et al.’s cell construction is difficult to follow. First: they present a diagram of how charges and dipoles supposedly align at the surface of a metal in contact with their electrolyte. The argument goes that everything necessarily aligns so that a layer with a (permanent?) negative capacitance forms near the surface of the metal.
The authors then connect this, somehow, to two stable states for the electrode itself (either the state of being Al metal, or being Li metal). How this connection between stable states of the electrolyte and stable states of the electrode comes about is not clear — I would say they are not related at all. The clearest indication I have as to where this falls down is here, where they describe how “self-cycling” starts:
As the self-cycling process starts, triggered by a critical thickness of the new plated phase on the negative electrode, the electrode’s Fermi level switches from µAl to µLi, forcing the movement of additional cations to the interface Fig. 1(b). Two electric fields with opposite directions are then applied to the mobile cations and, as the electric field due to the trapped negative charges E0 surpasses the electric field due to the electrode electrons E, the current reverses and discharge starts ( E’ > E => Ceq < 0)
The authors seem to suggest here that a situation arises where the total capacitance in the system becomes negative (Ceq < 0), which in theory should charge itself when it is nominally discharged. Even if this was the case, this provides no explanation at all for any conversion of energy from outside the cell. Any ‘self-charge’ would have to be a very local effect (i.e., at the interface only, for very short times). To have the overall cell charging itself, with no external input of energy, clearly breaks the laws of thermodynamics. I find it very challenging to follow the precise thinking here, but I believe it is grounded in much the same issue of believing that energy levels are fixed in certain positions that I discussed in my Part II post. But at some point it becomes too hard work to try and dig into yet more contrived theory, so let’s look at some experimental issues instead.
How not to detect Li metal plating
Now then. As we just saw, Braga et al argue that a self-cycling process arises because lithium metal (Li) is plated onto an aluminium (Al) electrode - and the cell finds itself in a situation where atoms or whatever suddenly snap back like a spring, charging itself to revert to an earlier state. The authors try to support this interpretation by proving the existence of Li metal deposition by x-ray photoelectron spectroscopy (XPS). XPS is a technique which allows identification of elements and, importantly, their local chemical environment (depending on the precise detected energy of emitted electrons) existing at the very extreme surfaces of materials (i.e., the top few nm of a sample - this is key, as we will see shortly). With this technique, they find a peak in the XPS spectrum which can be clearly identified as coming from Li, and comparing the position (binding energy) of this peak with a previously reported value, identify it as Li deposited on Al:
Li deposited on Al, Li/Al, was observed to show a Li 1s peak equal to 55.40 eV (Ref. 52) which coincides with the lithium peak observed in this study and shown in Fig. 3.
There are some problems with this. The first — and somewhat important — problem is that Li does not deposit as the metal on Al, it alloys with it, forming a range of Li-Al alloy compounds. The reaction of the two metals is spontaneous, so any sort of Li film deposited on Al would not last long.
The second is that Li metal and most of its compounds give peaks in the XPS spectra which fall in a very narrow range of binding energies. Braga et al. actually demonstrate this by listing a range of common Li-containing compounds, all falling between 54 and 56 eV. Immediately below though, is Figure 3, which shows the Li 1s XPS spectrum with seemingly only one broad peak between around 53 and 57 eV.
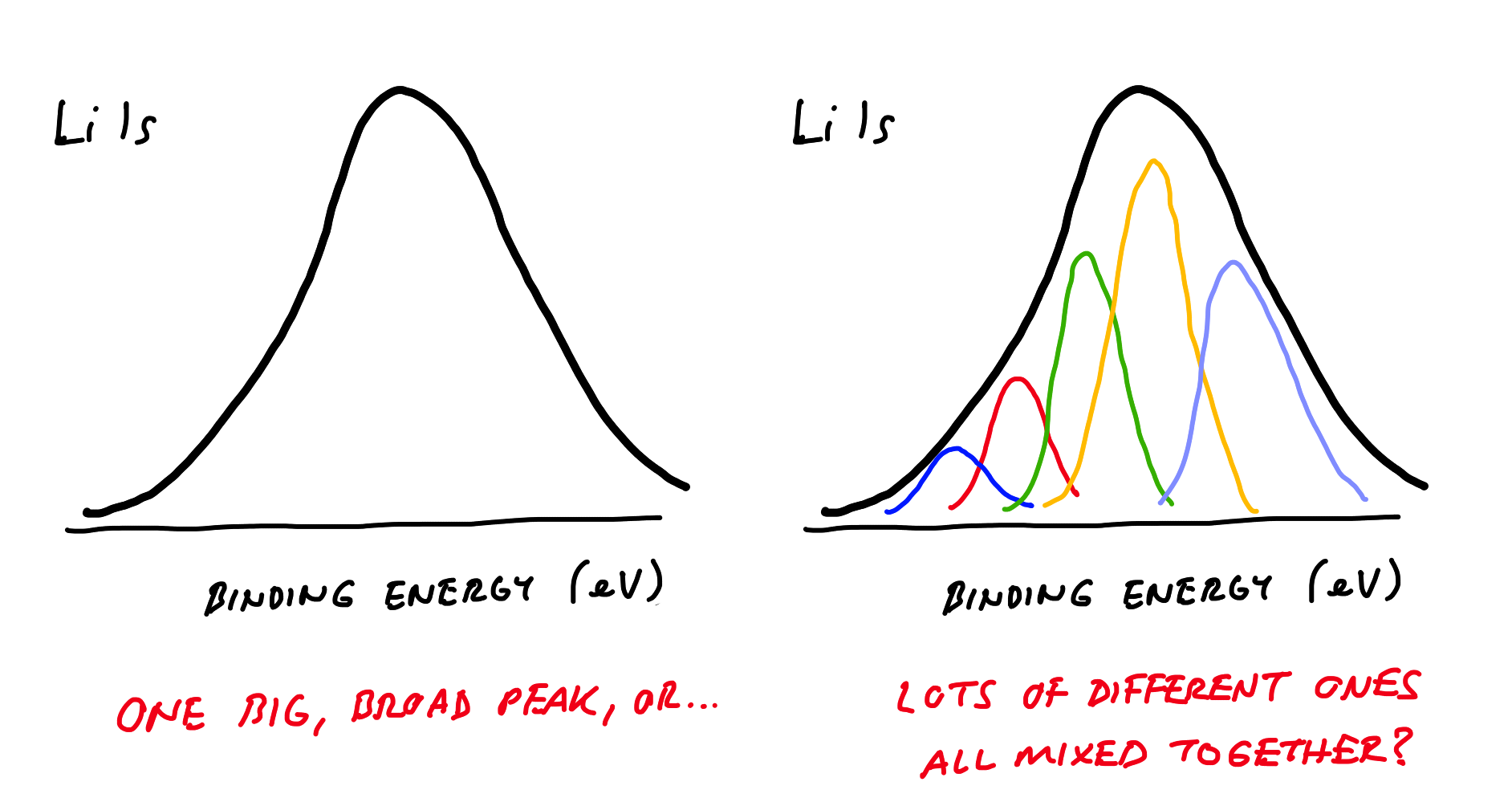
So what does this mean? Well, it means that with the peaks from all the possible compounds being so close to each other, it is impossible to know in this case if the experimentally-obtained peak is a single compound or a mix of different ones — they all overlap, so it is effectively impossible to identify any particular Li compound (including the metal itself). This is not just an issue with this specific example - it is well known to XPS spectroscopists that so-called deconvolution of Li XPS spectra is difficult and generally unreliable for this reason. In effect, it just says that Li is at the surface, in some form or another. This is not surprising, because any number of reactions may have taken place to deposit Li compounds on the electrode.
What strikes me though, is that the authors considered that they would be able to detect metallic Li at all, given the description of the sample preparation. For reasons unclear to me — baffling in fact — the authors decided to seal their cells in epoxy resin, which they then had to “crush open”, in air, to get the electrodes out — which they then took into an Ar-filled glove box, to protect it from the air (?!). I would expect that most who have studied chemistry even at high school or similar know that Li metal reacts in air, forming a number of different compounds on the surface. Braga et al. even acknowledge this themselves, stating shortly after the previous quote:
The oxygen immediately reacted with the plated lithium on the surfaces of the electrodes
I don’t understand, then, why they even bothered with the glove box. Given that XPS only gives information from the very outer surfaces of samples, how can they expect to measure pure metallic Li? There is no chance that if a Li metal sample is exposed to air for any length of time, that any of the signal observed in the XPS comes from metallic Li itself. Yet, the authors conclude that:
It was shown by chemical analysis that Li plates on the negative electrode of an Al/90 wt. % Li -glass + 10 wt. % Li2S/Cu while charging. The amount of plated Li is sufficiently large to be clearly detected in XPS measurements.
Once again, it is hard to believe how such a basic and significant error makes it through peer review unchallenged.
Why should a small, low power electrochemical cell overpower an expensive, purpose built scientific instrument’s primary function?
An important point I want to pick up on is the question of how the authors provide evidence that their cells are “self-charging”, or “self-cycling”, and what we can read into this. I will start by focusing on the “self-charge”, since this is easiest to understand, and which we have already touched on.
The ability of an electrochemical cell to supposedly “self-charge” is central to this paper, and has been a theme in a few of the previous papers by these authors. Although it is rarely spelled out explicitly, the implication in “self-charge” is that it is exactly what it sounds like — that a cell has an ability to extract energy from its surroundings, convert it into electricity, and recharge itself. Therefore, even if the cell could only do this slowly and while in operation, if a battery could really do this it could extend its discharge time and, in effect, its energy density.
Self-charge, unsurprisingly, is rather unprecedented in batteries. But I can start to explain what self-charge could conceivably look like by discussing its opposite, better-understood phenomenon of self-discharge. Self-discharge is seen in many battery systems, where an unwanted, so-called “parasitic” reaction takes place between an electrode and something in the electrolyte, using the stored charge and energy in the cell to drive itself. Prominent examples include shuttling reactions from oxygen or ions such as nitrate in lead-acid, nickel-cadmium and nickel-metal hydride batteries, as well as the polysulfide redox shuttle in lithium-sulfur batteries.
Regardless of the precise chemical cause, a self-discharge reaction effectively means that a small electrical current is passing inside the battery instead of through the external circuit — and so the cell gradually loses its stored energy, which is wasted largely as heat. How much energy and how fast depends on the precise chemical cause.
We can see the effect of self-discharge in a number of ways. Often, the self-discharge means that a battery requires passing more charge (charge = current x time) to fully charge it than to discharge it — the ratio of discharge to charge capacity is called the coulombic efficiency, and gives us an indication of how much of a self-discharge effect there might be. We can also charge the battery, wait a while, and then discharge it and see how much charge it has lost in the meantime.
We might then expect that a self-charge process should look similar, but opposite: some process converts energy from outside the cell — say, heat — into electricity, recharging one or both electrodes in the process, without extra current having to flow from an electrical power source. We could expect to see a cell that takes less time to fully recharge, than it does to discharge at the same current.
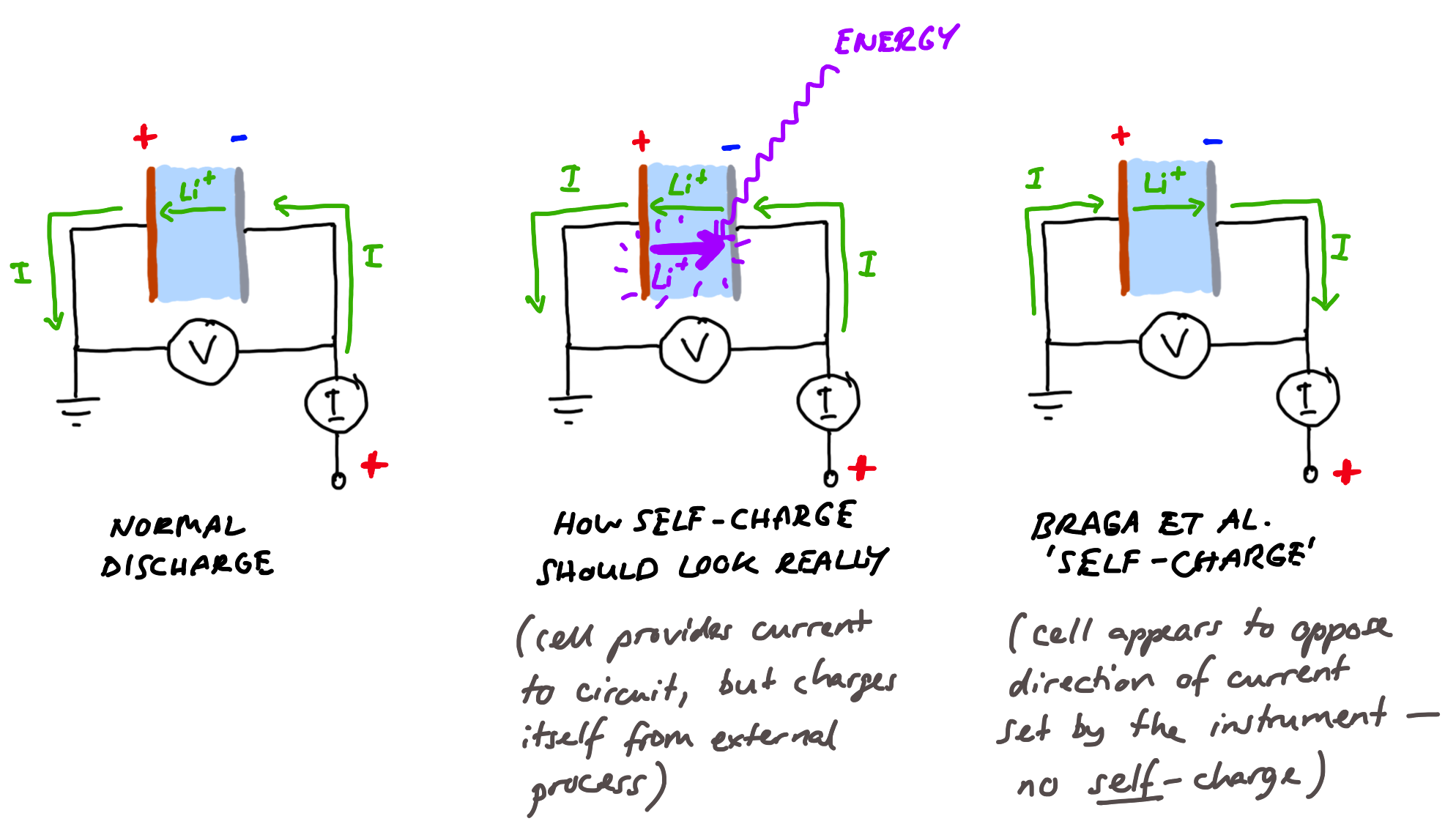
Do Braga et al.’s results show this? Well, no. In both tests where cells are tested with a constant current discharge (by convention, a negative current), the authors point out that self-charge is evidenced by measuring a positive (charging) current. That is, a different current, flowing in the external circuit.
We highlighted that the overall measured current remains positive even if the cell is set to discharge by setting a negative control current, Icon.
This is, then, not at all what we should expect. The authors argue that the processes happening inside the cell in some way force their testing instrument (a potentiostat/galvanostat — an instrument designed to apply controlled potentials or currents to electrochemical cell) to pass a different current than the one the instrument is trying to apply. The instrument is supposed to discharge the cell with a constant current of -1 µA, but instead measures a current of a few µA flowing in the other direction (i.e., charging), which is also fluctuating up and down (that’s the “self-cycling” part).
…the potentiostat cannot reverse the control current to positive when set to a discharge. The measured current is positive (in the opposite direction to the control current) and is entirely due to phenomena occurring in the cell that overcomes the control current.
The authors argue that it is physically not possible for the instrument to apply a positive (charging) current when it is programmed to apply a negative (discharge) current, because the instrument needs to be fixed in a configuration where it can only do one or the other. Therefore, the argument goes, if you tell the instrument to discharge, but it measures a charging current, it is the cell that is forcing this charging current through the circuit somehow. This is extraordinarily unlikely. The instruments which the authors are using here, which test one cell at a time and cost in the range of 10,000 — 20,000 EUR each, are purpose built to pass whatever current or apply whatever potential the operator requires with a very good accuracy, and to very quickly react to any changes in the cell that might happen that might affect this — so long as it is used correctly, and the operator understands its limits. They have a compliance well in excess of the voltage of these cells, so that they have the capability to apply whatever voltages they need to to make sure that the current measured through the circuit is what is asked for, to within a fraction of a percent.
What they are not, however, is foolproof. In the hands of an inexperienced operator, with the wrong instrument settings, connected up incorrectly, bad contacts, or just if some component on the instrument is broken, they can behave unpredictably and give spurious results. There is one particularly strong indication that this is the case here, in the Supporting Information: the authors discharge a commercially available button cell battery with the same low current, and although it does not appear to oscillate significantly, the instrument measures a significantly different current than the applied current, outside of the specification of the instrument.
It would be easy to test whether this behaviour is a true effect of the cell or not. Try the same thing with different settings, perhaps, and different instruments. To be fair to the authors, they have shown results from different instruments and argue this is not an effect of a broken instrument. However, every experiment is very different, there is never any consistency or systematic experimental design - and so this proves nothing. For all we know, the reason the experiments are so different is because they are not repeatable, and these are just those which gave the “self-charging” result. It is certainly the case that the experiments do not look as if they were designed to test for “self-charging”.
Have oscillating systems in electrochemical systems really never been seen before?
I want to highlight one final, technical point, which I find truly astonishing. It has been a common theme in this wider “glass battery” development to make sweeping and lofty claims, but I was startled all the same to see the clear statement in this paper that oscillations in electrochemical systems have not previously been reported “or anticipated”.
The reality is that electrochemical systems which oscillate — for whatever specific reason — are well known and have been studied for decades. Anyone can learn this by making a basic Google Scholar search. Really, the only way Braga et al. could be under the impression that they have not been seen before is because they did not look.
Considering the authors seem to want to give the impression that they are breaking new ground by forging new links between different disciplines, it is unbelievable to me that they seem not to have tried to learn from the field of electrochemistry — ironically, the field that gave birth to batteries in the first place.
What if I’m wrong, and the theory’s right?
Ok — let’s say for the sake of argument that their interpretation of the results turns out to be right, and they really have this remarkable electrolyte which makes self-charging batteries possible. How significant would these results be, in terms of the technology we have now?
If you compare where battery technology is today — I’m afraid the answer is “not at all”. Modern Li-ion battery electrodes can store a charge equivalent to around 3 mAh per sq cm of electrode area (mAh/cm2), can be easily charged in 1 hour (at a current of 3 mA/cm2), and cycled hundreds if not thousands of times. Even many academic researchers in the battery field, I believe, are unaware of how high this bar actually is.
In many of the experiments here, Braga et al. are discharging their cells, which have an area of 6.25 cm2, with a current of 1 µA - that’s 0.00016 mA/cm2. If you tried to charge a modern Li-ion battery, like the sort you find in almost any device now, with that level of current (scaled appropriately), it would take you more than two years to charge it from empty to full. These are miniscule currents, miles away from any current energy storage technology. The supposed self-charging effect, even if it were to exist, is only shown to be up to a few µA, and since we have no evidence that it scales, I have no reason to believe it could ever be put to practical use. None of the performance figures are remotely useful if you look at them objectively — none at all.
And what about the energy which is converted in order to drive this self-charging reaction — where does that come from? In this paper, the authors never directly say so. We get a hint early in the introduction:
A high-priority technical target today is to harvest energy coming to earth daily from sunlight and wind energy, but there is no known way to harvest, clearly and efficiently, energy from waste heat.
This is not expanded on in the rest of the paper. Only in comments made to the media, and in this patent entitled “Heat energy-powered electrochemical cells” do the authors reveal that are claiming that their cells are self-charging by directly converting heat to electricity. The theory does not explain this. But more to the point, even if it existed, I don’t believe the effect could even be measurable in these experiments.
If the glass is converting heat into electricity — say, its own heat — I would expect that it should cool down. I estimate, based on the size of the cells given, that the authors have about 1.4 g of their electrolyte in each cell. The maximum “self-charging” current in these experiments we see is around 5 µA. At a cell voltage around 3 V, that’s a power of 15 µW. Now, we can assume that the heat capacity of the electrolyte is around 1 J per gram per °C — that’s about what it is for most batteries. This says that it takes 1 J (1 W for 1 s) to raise the temperature of 1 g of material by 1 °C. In this case, that means converting heat and completely turning it into 5 µA of current at 3 V would need 1.4 J/1.5 µJ/s = 93000 seconds, or about 26 hours, to cool the glass by 1 °C. Without it being warmed back up by anything in the surroundings in the meantime. If this was at all detectable, I would be amazed.
Numbers are important.
Summary
I have followed this line of work for three years now. Initially, I found the whole thing curious — instinctively I knew that the eye-popping claims of an 8,000 Wh/kg battery that simply pulled Li metal off one side of an esoteric electrolyte and deposited essentially the same Li metal back on the other side had to be impossible.
But with so much vague and conflicting information, it was hard to put together a coherent explanation for what was really happening. Others preferred to focus on pointing out where the theory that the authors were proposing was violating the laws of thermodynamics. However, I got the feeling that many readers who are not experts in this scientific area felt they probably couldn’t judge this for themselves, took the main performance claims at face value and concluded that “it looks like it works, but we don’t know why”. My aim with these posts has been to convey the message that at first glance it might look like it works, but it wouldn’t in practice — because there’s no way you can trust these results.
I figured, perhaps naively, that the results soon prove to be unreplicable, and a strongly skeptical response from the battery community would put the brakes on this work a little bit. That didn’t happen, at least straight away, and it is yet to be seen if it will happen. So more work got published, I was interested in where this was going, thinking that this had to collide with reality fairly quickly, and people asked me questions about it — so I read, and sometimes wrote…
Over three years some of my initial hunches have been borne out, though not necessarily in the way I had thought. Some of this is thanks to excellent work on the part of other scientists, such as the TU Graz team mentioned above — but some of this has come from the authors themselves. With nine papers to read, as well as numerous media interviews, some of the key arguments have popped up in enough places and in enough forms that one can start to follow the thinking a lot more clearly, and spot the mistakes a lot more clearly.
And after three years I feel quite comfortable in making my conclusion: this body of work bears all the hallmarks of pathological science. From the beginning, the clear objective has been the development of supposedly groundbreaking battery materials that would change how we think about energy storage research. These are all very hard papers to read — they do not give the impression that they putting much focus on explaining their theory and results so that readers understand as best as possible.
Every result is viewed from the perspective that the glass electrolyte material is essentially perfect — defect free, super-conductive, stable in all relevant conditions, with no unwanted side-reactions to speak of — even though there’s no real evidence they know what the electrolyte is. Materials are assumed to be free of well-known havoc-wreaking impurities like water and oxygen, even though they are handled extensively in air. For every unexpected result, a previously unanticipated aspect of materials physics theory has to be constructed to explain it, and no unwanted degradation reaction — generally the bane of any other new battery material — or some artefact of the experimental method can be considered as a sufficent explanation instead.
Experiments are presented without any sense that they constitute a logical experimental matrix to test for the effect of a particular variable — rather that they are expediently presented to “confirm” a particular aspect of contrived theory. Several key claims, such as the observation of the supposed “self charge” phenomenon, were first communicated to the world through media interviews rather than a scientific paper. In several cases, some of these papers made references to results in unpublished work, and in one case even in a review paper — which is certainly the first time I ever saw a review of work that hadn’t yet been published. Isolated performance characteristics, measured in tiny devices passing tiny currents, are linearly extrapolated into completely unrealistic figures for hypothetical real devices, and passed on entirely uncritically to an unsuspecting general public, which has an increasing interest in new battery breakthroughs. I could go on.
I don’t think I have met that many people in the battery field in the last three years who didn’t immediately recognise that this was flawed work that is, in most respects, a dead end, although most would not say so openly. So it is with some bemusement when I read about Hydro-Québec’s recent announcement that they intend to start work on commercialising the patents related to this work. I do not know how deep Hydro-Québec’s interest really is in this. But much of the press that accompanied this press release (e.g., here, here) repeat some the aforementioned performance claims — claims that are completely unsupportable. I don’t know what the result will be of Hydro-Québec’s project in the two years they will supposedly work on this, but I will happily take bets on it not being a super-high-energy, super-fast-charging Li3OCl-based glass battery with a lifetime of 23,000 cycles. I may renew my earlier guess that it will quietly disappear.
A final note
I of course realise that with this post I am strongly criticising work which has a recent Nobel Prize winner in John Goodenough as a major contributor, something which might be thought politically unwise. I, naturally, hold the achievements for which he won the Prize in the highest regard. But it should not be controversial to make the scientific observation that in this case, he and his co-authors are way off the mark.
Some readers may be of the opinion that in light of John Goodenough’s achievements and track record, that he must be onto something and we’re all missing something. First, I will point out that argumentum ab auctoritate is a logical fallacy; smarter men than me have been wrong before. Second, I have made every effort to explain the authors’ results as honestly as I can and my perspective as clearly as I can. Comments are open here, and if someone thinks I have made a mistake and can show it clearly — then by all means, please do.
And lastly — some readers may consider it inappropriate to place such criticism “in the open” such as this. I don’t have an academic job anymore: I write this in a private capacity, purely out of personal interest. I have no incentive to bury this in some paywalled “peer-reviewed” journal. But more to the point — in my view, any scientific research which receives and seeks public exposure in the way this has — never mind public funding — does not have a valid claim to immunity from public criticism. I have written this post precisely because of the public exposure this work has had. I don’t think it’s fair to argue that we can have praises sung of research papers in the press but criticism must be quietly and tediously submitted in the “proper” channels. I have written this post precisely because I see it, in part, as a unusually prominent and extreme manifestation of many issues we have with scientific integrity in the battery research field (but that’s another discussion, and maybe another post). We scientists need to be able to say openly “we can and should do better than this”. Especially of our role models with Nobel Prizes.