The most recent post published on this topic is Part III, published on March 13th, 2020. You can find that post here.
Read my previous post on the Energy Environ. Sci. (2017) paper here. I’m sorry, this one’s really long too (~5,000 words).
Glass battery, part two: the saga continues
Those following the story of the “glass battery” work by Maria Helena Braga and the battery world’s most high-profile figure, John Goodenough, may well have noticed the report of a new battery system in a paper entitled “Nontraditional, Safe, High Voltage Rechargeable Cells of Long Cycle Life” in the Journal of the American Chemical Society in April 2018. This new battery system is based around the same “glassy” electrolyte presented in their earlier work in a 2017 issue of Energy and Environmental Science. That earlier work attracted plenty of skepticism from other battery scientists, not least me: I wrote a rather long post detailing my own considerable objections to the claims in that work.
This new paper, however, presents some new startling claims of its own. For example: we now have a claimed lifetime of 23,000 cycles, a capacity which seems to increase over time without a clear limit, and an ability to “self-charge”. These properties, much with the huge energy density claims in the previous work, would be revolutionary if they could be reproduced in devices of the scale needed for, say, electric vehicles. Steve Levine at Axios has already reported on some of the skepticism to some of these claims, but as was the case with essentially all of the press on the previous paper last year, there has been no real analysis from a scientific point of view.
Gerbrand Ceder was quoted in that article as saying that this new work is “not what it is stated to be. Most of us have moved on from this saga.” I do agree, but the problem is that Goodenough has a high enough reputation that the majority of people not in the know (i.e., the general public, potential funders, investors) will implicity trust that he really is onto something and “the skeptics” (e.g., me, and most other scientists with some experience in this area) maybe don’t understand some new science, or are just wrong. It’s fallacious reasoning - but the rest of the battery field cannot expect the general public to take “the skeptics” seriously without trying to explain what we find problematic in these works. Otherwise we might as well look like those who doubted people like Galileo or the Wright brothers. And it does not help matters that this new paper, unlike the last one, is paywalled.
The new battery - it’s not that “nontraditional”, and it’s not “all-solid-state”
This new paper is, I think, rather more complicated than the 2017 one, but let’s start with the basics: the construction of the battery.
Despite being described as “nontraditional” in the title of their paper, the battery chemistry Braga and Goodenough are reporting here is actually rather “traditional” compared to their 2017 work. Gone, essentially without explanation, is the controversial claim of extensive plating of lithium metal on the positive electrode at potentials far more positive than the thermodynamically expected potential; the positive electrode is now a relatively conventional lithium insertion electrode with the formula Li[LixNi0.5−yMn1.5−z]O4−x−δFx.
It isn’t explained why this particular compound was chosen, but it is similar to a well-studied high voltage electrode material known as LNMO (LiNi0.5Mn1.5O4). The paper provides data for this material when made into a very traditional-looking electrode with carbon conductive additive and PVdF binder, then made into a traditional-looking cell paired a lithium metal negative electrode and a conventional liquid electrolyte - and it looks as I would expect it to, so there’s nothing controversial there.
Then, in comes the “glass electrolyte”. This is made in essentially the same way as in their 2017 work, so my doubts about its composition and behaviour as I detailed in my previous post still stand - I won’t repeat these here. However, to make contact between the electrode and the electrolyte, the authors coat a relatively large amount (30% by weight) of a waxy electrolyte (“plasticizer”) made from succinonitrile onto the electrode, and then they add “half a drop” of a liquid electrolyte to make the electrode and electrolyte stick.
It’s impossible to know how much “half a drop” really is, but it is probably a very large amount compared to the amount of active material (the LNMO-like compound) on the electrode. The amount of electrode material used in their cells is teeny-tiny - stated to be only up to 0.35 mg cm-2, which gives their “traditional” liquid electrolyte cells a capacity of ~0.06 mAh per square centimeter of electrode area. Electrodes in “traditional” lithium ion batteries have around 2 mAh cm-2, or more. In such batteries the weight of the electrolyte is often maybe 30% of the weight of the active material, but “half a drop” might be 10 mg or more, which would then be maybe five times the weight of the active material in the cell. There are therefore three different electrolyte materials at the positive electrode/electrolyte interface. This complicates things a lot, and I will come back to this later.
In any case, it is simply false to describe the cell as “all-solid-state”, as the authors do repeatedly.
So this battery increases in capacity over time? How?
Before I get to why the capacity might be increasing I have to back up to discuss an experiment which is presented in the paper but mostly skipped over. I’ve drawn below simple schematics of three different cell constructions which are presented. The “traditional” cell with the liquid electrolyte and the “non-traditional” cell with the waxy succinonitrile plasticiser and the “glass” electrolyte I have already mentioned. But, the authors also briefly tested a cell with an electrode containing the plasticiser along with a liquid electrolyte as an intermediate experiment.
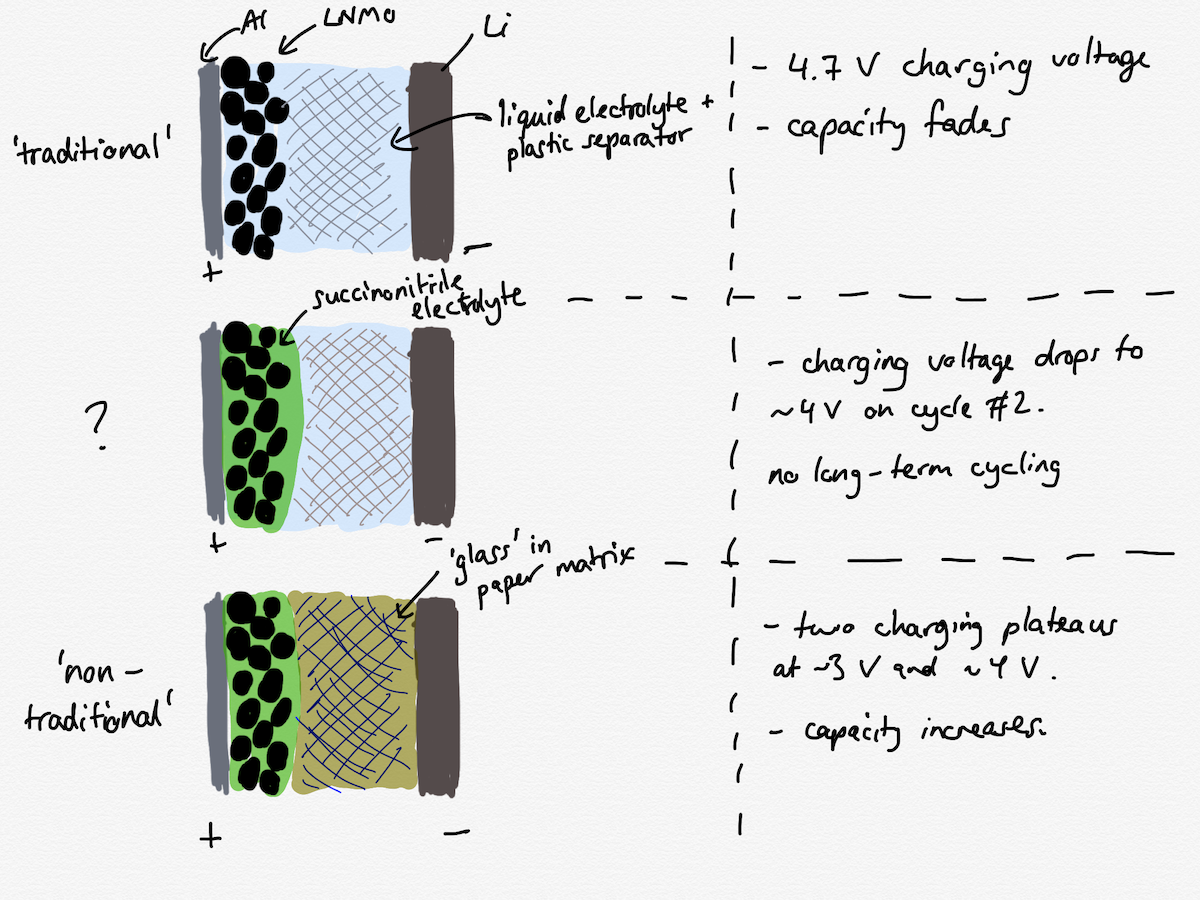
The discussion about the properties of the succinonitrile component is not particularly detailed and, I think, mostly wrong. The conductivity of the succinonitrile layer was determined (Figure 2a in the paper) by impedance spectroscopy (which I know plenty about) and the authors found it to be lower than they were expecting from the literature. In their experimental section, they say:
A mixture of succinonitrile (SN) plasticizer and LiClO4 salt in a molar ratio of 20:1 was heated in an argon-filled glovebox (MBraun, Germany) to get a transparent solution. The LiClO4 was mixed into the SN, but the conductivity of the SN did not increase as much as expected8 owing to not melting the LiClO4 prior to mixing it with SN.
The transparent solution tells me that the salt dissolves just fine in molten succinonitrile, so why they think that because they didn’t melt the salt as well is some sort of problem is beyond me. Actually, since LiClO4 is a strong (if relatively kinetically inert) oxidiser, I really would not recommend melting it and adding it to an also hot organic liquid because I’m not sure it wouldn’t just burst into flames.
Anyway, the authors present impedance measurements on the cells as well (Figure 2b) and conclude that the addition of the succinonitrile:
…creates a quasi-blocking electrode characterized by a Warburg impedance element associated with a charge-transfer resistance and a double-layer capacitance.
This isn’t supported by the results at all - there is no significant difference in the impedance of the cell with or without the plasticiser when using a liquid electrolyte - it is only when the “glass” electrolyte is introduced that the cells have a very high impedance (with internal resistances approaching 10 kΩ - close to 1,000 times higher than what I’d expect for most “traditional” batteries). Which is also an interesting observation in itself, considering the supposedly high conductivity of the “glass”.
Anyway, the telling result comes in Figure 4c, where the authors test the cell with the liquid electrolyte and the plasticiser. The cell initially behaves quite similarly to the “traditional” cell without the plasticiser on charging the cell (the cells are assembled in a discharged state), but on the first discharge it shows about half the capacity, and on the second charge, the charging voltage drops considerably, and no end to the charge is shown. All the authors say about this test is this (emphasis mine):
The electrochemical cell with the organic-liquid electrolyte contacting the anode and a plasticizer contacting the cathode (c) showed a rapid reduction of the charging voltage and no discharge current by the second cycle, indicating metallic lithium was not plated on the anode from the mobile cations in the liquid electrolyte.
The conclusion that metallic lithium is somehow not plated back onto the Li electrode at the negative electrode/electrolyte interface during charging is completely without supporting evidence, and assumes that the positive electrode/electrolyte interface is still working perfectly. But - and it is a really big but - in this comparison they have only changed the materials at the positive electrode and the negative electrode/electrolyte interface is in principle exactly the same! That is to say, the authors have changed the materials of the positive electrode and appear to be arguing that as a result they have changed the properties of the negative electrode instead. How this is effect is supposed to happen is left completely without discussion.
If I obtained this result in the lab, I would automatically work on the basis that the plasticiser layer I had added was not actually electrochemically stable in the operational window of the battery. It could be the succinonitrile oxidising, it could be the propylene carbonate in the “half a drop” that is oxidising, it could be corrosion of the aluminium current collector because of the lithium perchlorate salt - it could be all three.
The fact that the authors do not discuss this observation any further is inexplicable and in itself undermines the credibility of the rest of the article. If there are side reactions associated with the plasticiser layer, then these can be expected to still be there when the “glass” electrolyte is added, and this has to be taken into account in any discussion of the efficiency or cycle life of the cell.
So what’s going on in the “non-traditional cell”?
Compared to the 2017 work, there is a relatively large amount of data to consider, and to avoid any possible accusations of copyright infringement I would rather not reproduce plots here (my apologies to those who cannot access the article). So, I will summarise here some of my observations about the battery test data of the cells with the glass electrolyte before I talk about the authors’ analysis of the data.
Firstly, compared to the 2017 paper, data from five or six different cells is presented (there is additional data in the Supporting Information). The presentation of the data is somewhat inconsistent and the test conditions are a little different in each case, but all the cells seem to have (what I would say are) significant differences in their behaviour (voltage, capacity etc.). This suggests to me that these cells are not that well-reproducible.
There is a good example of this if one compares Figures 4f and 5c, which show how the “middle voltage” changes on repeated cycling. The cells tested in these two figures are apparently identical, but tested at different applied currents.
At a slower rate of 23 mA g-1 (Figure 4f), the “middle voltage” stays approximately constant at 3.37 V while the capacity gradually increases by a factor of five over the first 250 cycles or so. Over the next 60 cycles, the middle voltage drops, and the rate of capacity increase starts to slow down (only 308 cycles are shown). In Figure 5c, at a rate of 153 mA g-1, the “middle voltage” starts off closer to 2.5 V, increases to around 3.3 V over the next 2,000 cycles, then suddenly jumps up to 3.7 V where it remains more or less constant for the next 13,000 cycles. I don’t see that these observations are consistent and the authors don’t seem to connect them.
Secondly, a large part of the analysis and the discussion of the “self-charge” mechanism appears to hinge on the observation that the cell shows coulombic efficiency (the ratio of the discharge capacity to the charge capacity on any given cycle) in excess of 100% for “many cycles”. It is true, for many cycles the indicated coulombic efficiency is over 100%, but for the 15,000 cycle test I just mentioned, the coulombic efficiency for any given cycle usually falls anywhere in the range 95 - 105%, which I would say shows that the cell is “all over the place”. Any normal well-functioning lithium-ion cell would have a coulombic efficiency consistent to within a small fraction of a percent, which will mostly depend on what instrument is being used for the measurement and how stable the temperature in the room is.
What >100% coulombic efficiency should tell you
The only thing that a coulombic efficiency of more than 100% indicates is exactly what the definition suggests: that more charge is passed during the discharge of the cell within the specified voltage window than is passed during the charge of the cell.
All lithium-ion batteries can be expected to have a coulombic efficiency of slightly less than 100% on any given full charge/discharge cycle. That is, for the same charge/discharge current, the charging time of the cell will be slightly longer than the discharge. This is mostly because of unwanted reactions which happen especially when the cell is charging, such as the formation of a solid-electrolyte interphase (SEI) at the negative electrode or oxidation of the electrolyte at the positive electrode. Because of this, it is possible in theory to put more charge into the cell than it can theoretically take. This also tells you that you have to put more charge into the cell to charge it than you can get out when you discharge it, so this also impacts on the energy efficiency. However, in any well-functioning Li-ion battery, the coulombic efficiency is very close to 100% (e.g., 99.99..%).
By this definition, it is possible to have a coulombic efficiency more than 100%, but this would usually tell you that you have more side reactions on the discharge of the cell compared to the charge. For example, instead of oxidising the electrolyte, you could be reducing it: different side-reactions of course, but side-reactions all the same.
Obviously, this means that you could get more energy from discharging the battery than you put into it when you charge it. But this doesn’t violate thermodynamics: there is chemical energy stored in other materials besides the active materials, such as the electrolyte itself, which can be released if it is reduced (almost anything reacts with lithium to release energy). But, if this is the case, you will eventually run out of electrolyte (or lithium) to burn away and the cell will die. But, if there is a relatively large amount of electrolyte - like there often is in small test cells like these - this can take a long time. Many battery researchers are simply ignorant of the effect of electrolyte volume on the cycle life of small cells and this is part of the reason why claims of super-long cycle life in laboratory tests do not get repeated in the real world, where electrolyte volume has to be kept as low as possible to minimise dead weight.
But what is happening? Why is the capacity increasing?
Ok, ok - let’s get back to the article. I will pick out a few choice quotes to try and approach the authors’ argumentation, which is really not easy to get into. But bear with me…
Why the capacity of our cells increases with cycle number over an initial aging time appears to reflect primarily an increase with time in the dipole polarization contribution to the anode and Li+-glass electrolyte/plasticizer EDLCs.
The key word is “appears”. There does not seem to be any data to support this, and the cells already undergo “one month of aging” before the cycling begins, and the practical effect of this one month aging on the cell behaviour is not discussed or shown. And since this will become more important later - EDLC means “electrochemical (or electric) double-layer capacitance”.
The argument is essentially that double layer capacitance contributes to the charge storage of the whole cell, and that this capacitance can increase with time and therefore the capacity of the cell can increase with time. This is actually not unreasonable, but the problem is that DL capacitance is present in liquid electrolytes too and is very, very small compared to the capacity of the insertion electrodes themselves. Supercapacitors work on this principle, but they use extremely high surface area activated carbons as the electrodes. Even then, the energy they store is quite small compared to battery materials, which store charge in the bulk volume of a material and not just at its surface.
The origin of the reduced Coulombic efficiency in the first cycle of the all-solid-state cell (which may reflect structural changes in the active material) has not been totally determined, although it might be due to initial lack of polarization of the electrolyte and plasticizer and interface disorder. The subsequent steady increase in capacity with cycle number is typical of the self-charge capability due to spontaneous polarization of the Li+-glass ferroelectric electrolyte.5
Some more weak language with “may” and “might be”, but now we have the argument that the increase in capacity is in fact “typical” of the electrolyte’s “self-charge capability”. I didn’t remember this being mentioned in any of the previous work, but reference 5 is cited here. Wait, reference 5 is unpublished results?!?!
The increase of capacity, and therefore of the energy density, with increasing cycle number eventually gives a capacity that is greater than the theoretical capacity of the oxide host cathode particles. This extraordinary observation indicates that there must be a storage of charge in addition to that in the active particles.
This I can agree with. But…
The additional stored charge can only be electrostatic storage in an EDLC as in a supercapacitor.
No. As I have briefly mentioned here and also discussed in my previous post, there are a lot of possible side reactions which can contribute to charge storage in the cell. However, it is impossible to know, because I discussed several possibilities in the previous work and with the addition of the succinonitrile, liquid electrolyte and wider voltage window there are even more possibilities this time. In addition, the theoretical capacity of the cell based on the active material is so low now that it is very reasonable to expect that side reactions from the electrolytes - which make up the vast majority of the total material in the cell except for the cell housing - could dominate the behaviour. And it is entirely possible that the breakdown of the electrolyte can produce compounds which are redox active and effectively capable of storing charge on this scale. The authors simply do not seem to consider this.
What about the “self-charge”?
Before I get into this it’s worth pointing out that we got a hint about what Goodenough means by “self-charge” in an interview with Bloomberg in November 2017. In that, he said:
She [Braga]’s been lighting an LED with this battery charging itself for two years. It runs on ambient heat.
At the time this caught my attention especially because there was the direct claim that their battery was literally charging itself using heat from the atmosphere around it. There was no mention of this in any of their previous literature, and there is no battery known that can pull heat out of the atmosphere to charge itself without any applied current. This is a huge claim, and now that it’s actually in the published literature we can take a closer look at it.
As I noted previously, there is a mention of “typical” self-charge in relation to unpublished work, but there is a further discussion of the “nature of self-charge” in the Supporting Information. This discussion also cites a rebuttal to the formal comment on the 2017 paper published by Daniel Steingart and Venkat Viswanathan - a rebuttal which I would have expected to have been published at the same time as the comment but which I can find no trace of - so that’s a second unpublished work which they cite in relation to this supposed self-charge effect.
Anyway - this discussion of the “nature of self-charge” starts by describing one “self-charge” process as being common to “all kinds of battery cells”, which
happens due to the necessity to align each electrode’s Fermi level with the electrolyte’s Fermi level in contact with the electrodes at the electrode/electrolyte interface as demonstrated in ref. [S1].
(that ref [S1], incidentally is the rebuttal I can find no trace of).
This, as far as I can tell, is referring to bog-standard double layer capacitance, which is completely uncontroversial and has been standard theory in electrochemistry for more than a century. I have never heard of this being described as self-charge, certainly not the sort which charges your batteries for you while simultaneously cooling your apartment down. This double layer capacitance effect - the alignment of Fermi levels - occurs in every battery, on both electrodes - but there is no net change in the charge stored in the cell, so there is definitely no “self-charge”.
The discussion seems to continue to discuss this effect in the context of their solid electrolyte. The difference, they argue, is this:
…we argue that this self-charge is the result of equalization of the Fermi levels across the negative-electrode/electrolyte interface and a time lag between the arrival at the interface of the fast-moving electrolyte cation and the slower-moving electrolyte electric dipoles. Equalization of the Fermi levels by the interface EDLC (Fig. S9) is retained on arrival of the electric-dipole charge by a plating of an electrolyte cation onto the negative electrode, i.e. a self-charge, that creates a negative charge in the electrolyte.
Ok, this is complicated stuff and it’s not easy to read, but I think I have a handle on what they’re trying to say. They have diagrams which I also don’t think are that clear, so I’ve tried to draw my own to describe the interfaces formed between a Li electrode and a liquid or a solid electrolyte:
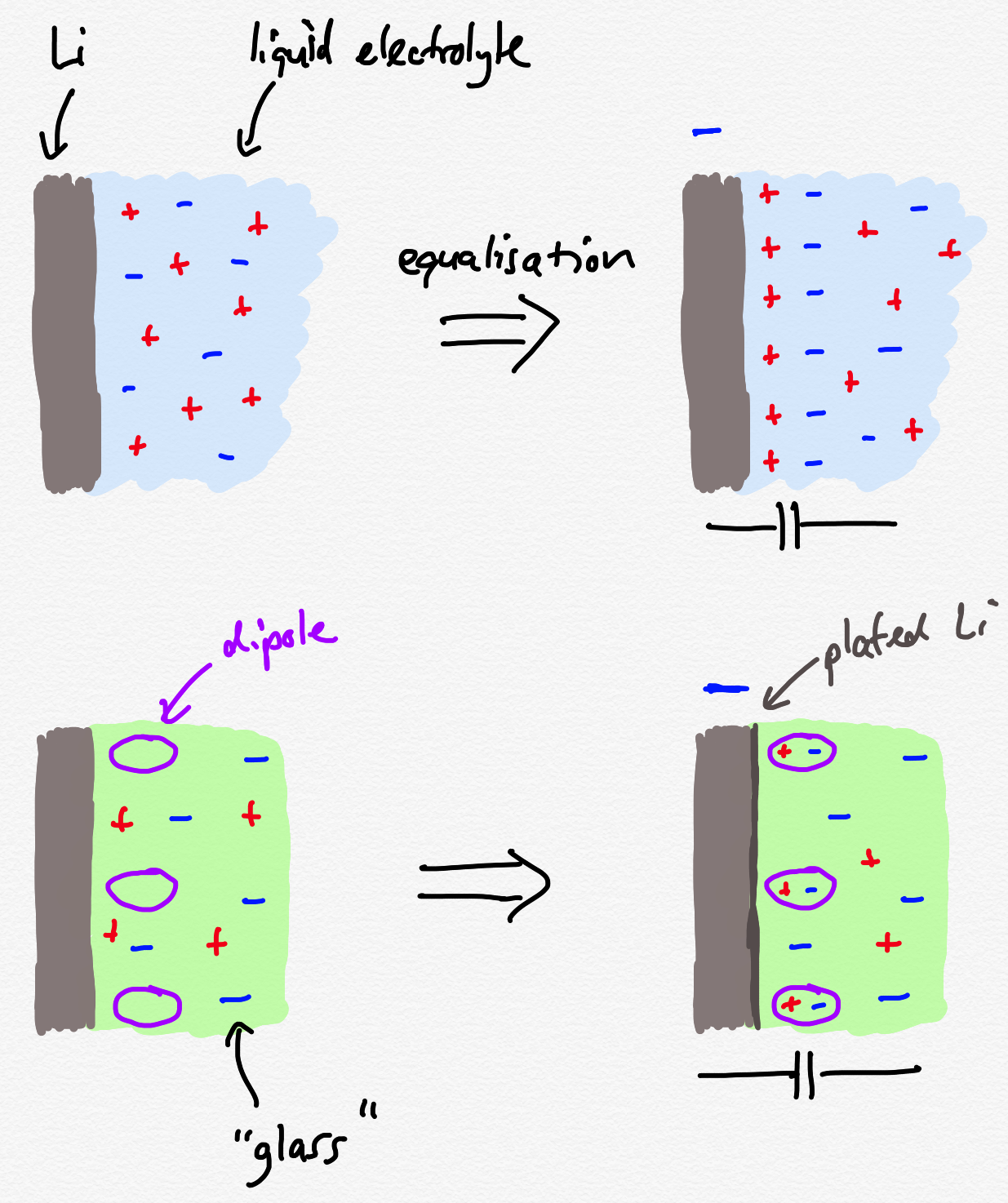
As the authors describe, the electrode and electrolyte have different potentials (Fermi levels in physics language) which causes charges to move in order to equalise this imbalance in energy. In a liquid electrolyte, both positive and negative charges (i.e., the ions in the electrolyte salt) can move, so they rearrange themselves at the interface so that positive charges form a layer along the nominally negatively charged Li surface. Then, the negative charges form a more loose layer on top of those positive charges. This is the “double layer”. The potential difference is then “dropped” across this double layer - effectively, the energy change as a result of this process is stored in the “double layer capacitor” that forms.
In an ideal solid electrolyte, only the positive charges can move. The negative charges cannot, because they are fixed in structure of the solid. But, there is still an energy imbalance that needs to be corrected. The authors argue that to do this, lithium can be deposited out of the electrolyte and dipoles in the electrolyte can become polarised in order to correct for the charge imbalance. I think this is fine, because it still creates a double layer in my view, but this only needs roughly one monolayer of lithium to equalise the energy difference - and this is where I think the authors have got it wrong - for much the same reasons they did in their 2017 work.
Moreover since plating creates negative charge in the electrolyte on the anode side, the electric field in the direction of E’’’ increases again and more cations are attracted to the anode surface while the cation-deficiencies diffuse towards the cathode charging the electrolyte/plasticizer EDLC. Furthermore, charging the electrolyte negatively makes the Fermi level of the electrolyte increase, discharging the anode/electrolyte EDLC and charging the electrolyte/plasticizer EDLC, which will discharge during cell’s discharge and contribute with the corresponding extra capacity.
They seem to be arguing that because the negative charges do not move, the difference in energy is never equalised, it just ends up somehow increasing without any suggestion of a limit - lithium ends up getting dragged out of the positive electrode, through the electrolyte, and onto the negative electrode, which represents an actual “self-charge” of the cell. And that this happens no matter what you do - they do say that this process happens on discharge as well as charge.
As they say in the article, charge is conserved in the cell. But - energy can’t be. Assuming this is all true, if you discharge the cell only at the same rate it self-charges, then you can draw energy out of it forever - you have a perpetual motion machine. So I’m pretty sure we’re back at breaking the laws of thermodynamics all over again.
The authors do actually suggest though that there is a limit to all this, bizarrely at the end of the introduction:
The cell capacity increases with cycle number until it stabilizes (not shown) at a value higher than the theoretical capacity of the cathode host oxide.
“Not shown” I think sums up this work.
Wrap-up
If I have been a bit more heavy handed in my analysis of this paper compared to the 2017 Energy Environ. Sci. work, it is because I am dismayed that papers on this topic have continued to be published from this group and continue to receive press exposure with very little serious critique, and moreover that none of the authors have made a substantial effort to engage constructively with “the skeptics”. Instead, we are waved away, laughed off or even criticised for “pouring cold water” on the work and discouraging those who might invest or obtain a license for their patent. I have to question their priorities.
Goodenough is on record as saying that “the skeptics do not understand the properties of an electrode/electrolyte heterojunction”, but without explaining exactly why the electrode/electrolyte heterojunction in these cells should behave so differently from any other similar system. It’s not as if their electrolyte is the only solid electrolyte - there is a huge literature on solid electrolytes and solid state ionics, not just in batteries but also other electrochemical devices such as solid oxide fuel cells. But the authors don’t engage with this either in this paper or their previous work.
And this, for me, is the biggest problem. In my previous post, I criticised the authors for not discussing essentially any of the wider literature in their 2017 paper, and this remains true now. It could be true that I don’t understand the properties of a heterojunction - but the authors of this work do not show much hint that they understand that much electrochemistry, because they show zero consideration of any of the myriad ways in which electrochemical cells can misbehave and give you unexpected results. And if there’s one thing I’m sure I understand, it’s that. (Otherwise I might need to find a new career!)
It is also a major problem, I believe, that some of the most controversial arguments in this new paper reference unpublished work, and we have seen how apparently key details are “not shown”. It is difficult to understand how any paper can make it through peer review on this basis, but this is not an isolated incident anymore - and this new paper is in the flagship journal of the American Chemical Society. What does this say about the editorial and peer review processes, not only of this journal but those in which their previous work on this electrolyte has been published?
I met up with a friend at a recent conference who explained to me that Goodenough is a great ideas man with a great grasp of physics, but he’s not an experimentalist - and so he trusts that when he is brought experimental data that it is correct, the cell is working properly and he has to be able to explain it somehow. I don’t how true this is. But it seems that no-one involved in the process of this work, from experimental design to publication, has given much consideration to any of the many ways in which the data might not show what they think it shows. And there is no reason - in my moderately humble but somewhat experienced opinion - for anyone to take any of these convoluted and unprecedented arguments seriously until they have proved beyond reasonable doubt that the chemistry of their cell is what they say it is.
As a final note: although I never received any direct questions or criticism on my decision to publish my previous post publicly on my own website rather than in any kind of peer-reviewed literature, I know others have been criticised for doing similar things (not just on this topic). To pre-empt any such questions: this research I’ve done in my spare time in the space of a couple of days, and I have no intention of wasting my time going through any sort of peer-review process, which I’m not confident is all that good a mark of quality anyway. I also do this because this is a direct response not just to the research itself but also the wide press it and the previous work has received - so I believe it’s important that questions are raised in the public domain. It just seems that there are very few people prepared to do this. I’m happy to hold up my hand and admit I’m wrong if presented with a well-supported argument, and correct anything in this post accordingly. I’m also happy to answer questions and try and clarify anything if requested. The comments are open below!